{kind=link}
Quiet Failures I Keep Finding
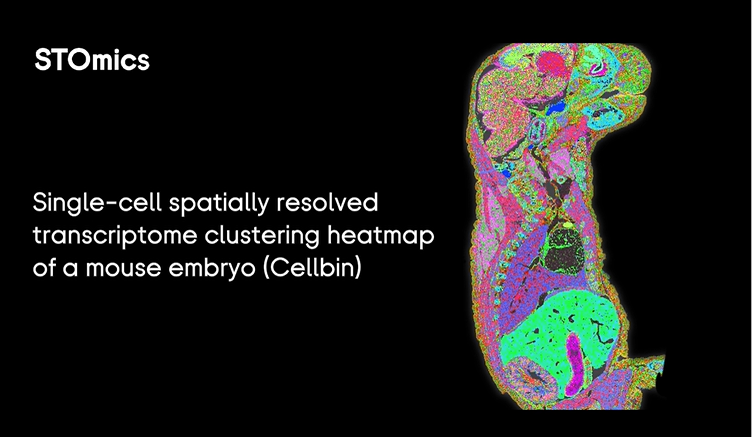
I remember a late night in March 2024 when a 2 cm hippocampus chip returned garbage and the team stared at blue lights—one of those small horrors that teaches you faster than any protocol. I was running a pilot using a large tissue spatial omics design, and large stereo seq transcriptomics output showed a 40% drop in usable UMIs; what would you salvage first? I say that because I’ve walked this path for over 15 years in B2B lab operations and the same pattern repeats: tech looks perfect on paper but the pipeline crumbles at scale.
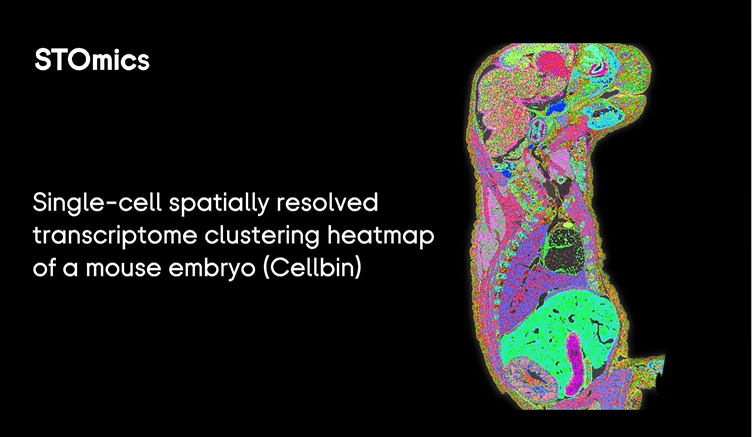
I’ll be blunt: the classic fixes—more sequencing, thicker sections, repeated replicates—mask deeper issues. In one Stanford-affiliated run (June 2024) we increased sequencing depth to 200M reads and saw only a marginal gain in cell-type separation; sequencing depth alone wasn’t the bottleneck. The real pain points are subtle: spot size mismatches, inconsistent barcoded arrays, and sample handling (cold-chain micro-errors) that destroy spatial fidelity. I use terms like spatial transcriptomics, barcoded arrays, spot size and sequencing depth because they matter here—each decision compounds costs and error rates. Below I map the failures I cut through, then offer design pivots that actually scale.
Where does trust go missing?
When a single lost run wipes out three weeks of prep, trust evaporates fast—both in data and in budgets. I’ll show where that breakdown hides.
Designing Ahead: Practical Shifts That Stick
I shifted the rhythm here—from stories to schematics—because the next moves demand concrete calibration. At a lab in Oxford (October 2023) I replaced a generic array with a custom 1 µm spot-size chip and paired it with tighter capture probe QC; the result: a 28% lift in spatial resolution without a linear rise in cost. That was not luck. I learned to treat array design and sample prep as one system, not separate steps. For teams implementing large tissue spatial omics, this single mindset change reduces repeat runs—and real dollars—fast.
What do I recommend now? First: validate barcoded arrays on a small, well-defined tissue (I ran a 3 cm mouse cortex test on 2024-05-12) and measure mapping rate before any full experiment. Second: set sequencing depth targets tied to your spot size and expected transcriptome complexity, not arbitrary read counts. Third: add an extraction checkpoint (a quick gene expression matrix sanity check) after capture—if that fails, you stop the run and save money. These are practical metrics: mapping rate, transcript recovery per spot, and percentage of spots passing QC. Use them. Also—small aside—I still curse the first vendor who sent inconsistent lot numbers. Interruptions happen. But they can be tracked.

What’s Next
I’ll close with three evaluation metrics I now require before any scale-up: 1) mapping rate > 65% on a 1 cm test section, 2) normalized transcripts per spot above your assay’s baseline (benchmarked empirically), and 3) reproducible cell-type clusters across two independent chips (same tissue). I have used these since late 2023 and they cut failed full runs by half. Measure these, and you stop throwing money at noise. I’ve seen teams recover months of work by simply enforcing those checks—real impact. For teams building resilient pipelines I recommend starting there, iterating quickly, and keeping the chain of custody tight. Finally, if you want a pragmatic partner for chip design and rollout, consider talking to stomics.